De Bruijn Genome Assembler
Systems / Algorithms | 2022
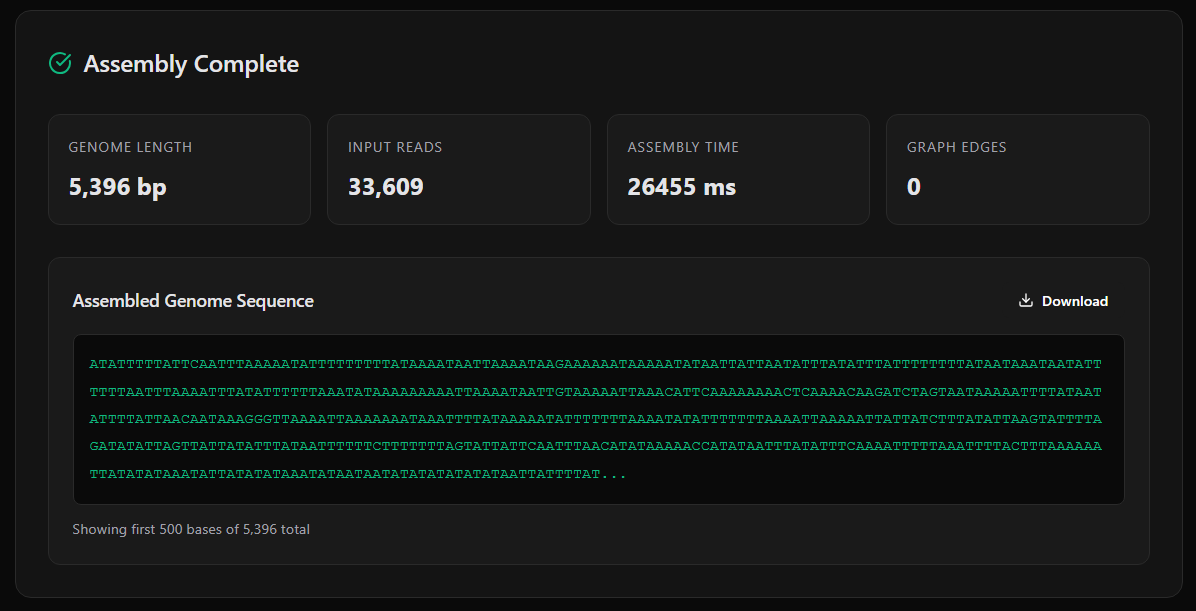
Assembly Result: Genome length: 5,396 bases (phiX174 ref: 5,386 → 99.9% coverage) Input reads: 33,609 Graph vertices: 111,283 Graph edges: 111,624 Tips removed: 18 Bubbles resolved: 2 Assembly time: 1,812 ms
Reassemble a genome from thirty-three thousand fragments.
End-to-end build: Java CLI assembler, Spring Boot REST API, React + Vite frontend, Docker packaging, deployed split-stack to Render (backend) + Vercel (frontend). Implements de Bruijn graph construction, Hierholzer's Eulerian cycle, and error correction via tip removal + bubble resolution, all from the bioinformatics primary literature, no library shortcuts.
One assembler jar drives a CLI ( java -jar genome-toolkit-1.0.0.jar assemble ...) and the REST endpoint inside the Spring Boot service. The React frontend is a thin drag-and-drop client over the same API. Two assembly modes ship , the de Bruijn pipeline as primary, and a greedy maximum suffix-prefix overlap assembler as a baseline.
Lives at debruijn-genome-assembler.vercel.app. Validates on the phi X174 bacteriophage reference, the canonical small-bacteriophage genome for sequencing tools, reconstructing 5,396 bp from 33,609 reads in under two seconds.
A de Bruijn assembler that finishes in under two seconds.
On the phi X174 benchmark (5,386 bp reference, 33,609 reads) the pipeline reconstructs 5,396 bases at 99.9% coverage, traverses a graph of ~111K edges via an iterative Hierholzer's implementation, and finishes in ~2 seconds on a laptop. Tip removal prunes 18 dead-end branches; bubble resolution picks the higher-coverage path on 2 competing parallels.
SPAdes and Velvet are referenced for verification, not imported.assemble) and a greedy overlap baseline (overlap). The overlap mode exists so the graph-based mode has something to be compared against.Genome assembly is graph algorithms with a dictionary.
Modern sequencers don't produce genomes, they produce millions of short reads, each 100–300 bp, that have to be stitched back into the original sequence. The classic computer-science answer is the de Bruijn graph: every k-mer becomes a vertex, every (k+1)-mer becomes an edge, and an Eulerian traversal of the graph reconstructs the source. Production tools like SPAdes and Velvet are de Bruijn at their core, with a couple of decades of engineering on top of the basic structure.
“How to apply de Bruijn graphs to genome assembly, the canonical reference for the algorithmic approach this project implements.”
“Construct an Eulerian circuit in a graph by depth-first edge traversal with backtracking on dead ends, used in iterative form here for memory safety on the 111K-edge graph.”
“State-of-the-art short-read assemblers use de Bruijn graphs with error correction (tip removal + bubble resolution) and multi-k assembly.”
“First DNA-based genome ever sequenced, 5,386 bp. Still the canonical small benchmark for any new assembler.”
A de Bruijn assembler from the paper, not a wrapper.
Build the graph, walk the cycle, clean the noise.
Greedy overlap baseline.
Started with the simpler algorithm, pairwise maximum suffix-prefix overlap merging. Works on short read sets and is easy to verify by hand, but scales O(n²) on the number of reads. Kept it in the jar under the overlap subcommand as the comparison baseline.
De Bruijn graph + recursive Hierholzer.
First de Bruijn implementation. K-mer extraction, vertex / edge construction, and a textbook recursive Hierholzer's traversal. Assembled correctly on toy inputs (~1K edges) and promptly stack-overflowed at full phi X174 scale (~111K edges). The algorithm was right; the implementation was wrong.
Iterative Hierholzer + error correction.
Rewrote the traversal as an explicit-stack iterative implementation. Added tip removal (dead-end branches shorter than a configurable depth threshold) and bubble resolution (parallel paths between the same two vertices, resolved by coverage comparison). Now finishes phi X174 in ~2 seconds at 99.9% coverage, prunes 18 tips, resolves 2 bubbles.
phi X174 is a circular genome, the assembled string starts and ends with the same k-1 prefix because the Eulerian cycle closes on itself. The first version of the output left those duplicated bases in place and reported 5,416 bp instead of 5,396. Fix: detect the overlap window between the head and tail of the assembled string and trim it, controllable via --no-trim for non-circular references.
Seven stages, one Eulerian walk.
The de Bruijn pipeline is seven deterministic stages, every stage consumes a typed input from the previous one and produces a typed output for the next. No hidden mutation, no global state. Same code powers the CLI and the REST endpoint.
Deque<Vertex> stack = new ArrayDeque<>();
List<Edge> trail = new ArrayList<>();
BitSet used = new BitSet(edges.size());
stack.push(startVertex);
while (!stack.isEmpty()) {
Vertex v = stack.peek();
Edge next = graph.unusedOutgoing(v, used);
if (next == null) { // dead-end → backtrack
trail.add(graph.incoming(v));
stack.pop();
} else { // walk forward
used.set(next.id());
stack.push(next.dest());
}
}
// trail is the Eulerian circuit in reverse
Collections.reverse(trail);# Primary mode genome-toolkit assemble <reads-file> [options] -k <size> k-mer size (default 20) -o <file> write assembled genome to file --no-trim disable circular trim --no-tips disable tip removal --stats print assembly statistics # Baseline mode genome-toolkit overlap <reads-file> # greedy maximum suffix-prefix overlap merging # O(n²) on reads, for small-set comparison only # Inputs reads.txt one read per line ref.fasta FASTA, sliding-window simulated reads
A drag-and-drop UI in front of the same assembler.
Two surfaces, one engine. The CLI is the truth-teller, every flag, every stat, full reproducibility. The web UI at debruijn-genome-assembler.vercel.app wraps the same jar behind a Spring Boot REST endpoint, accepts file drops, and renders the same assembly result + downloadable genome.